



版權說明:本文檔由用戶提供并上傳,收益歸屬內容提供方,若內容存在侵權,請進行舉報或認領
文檔簡介
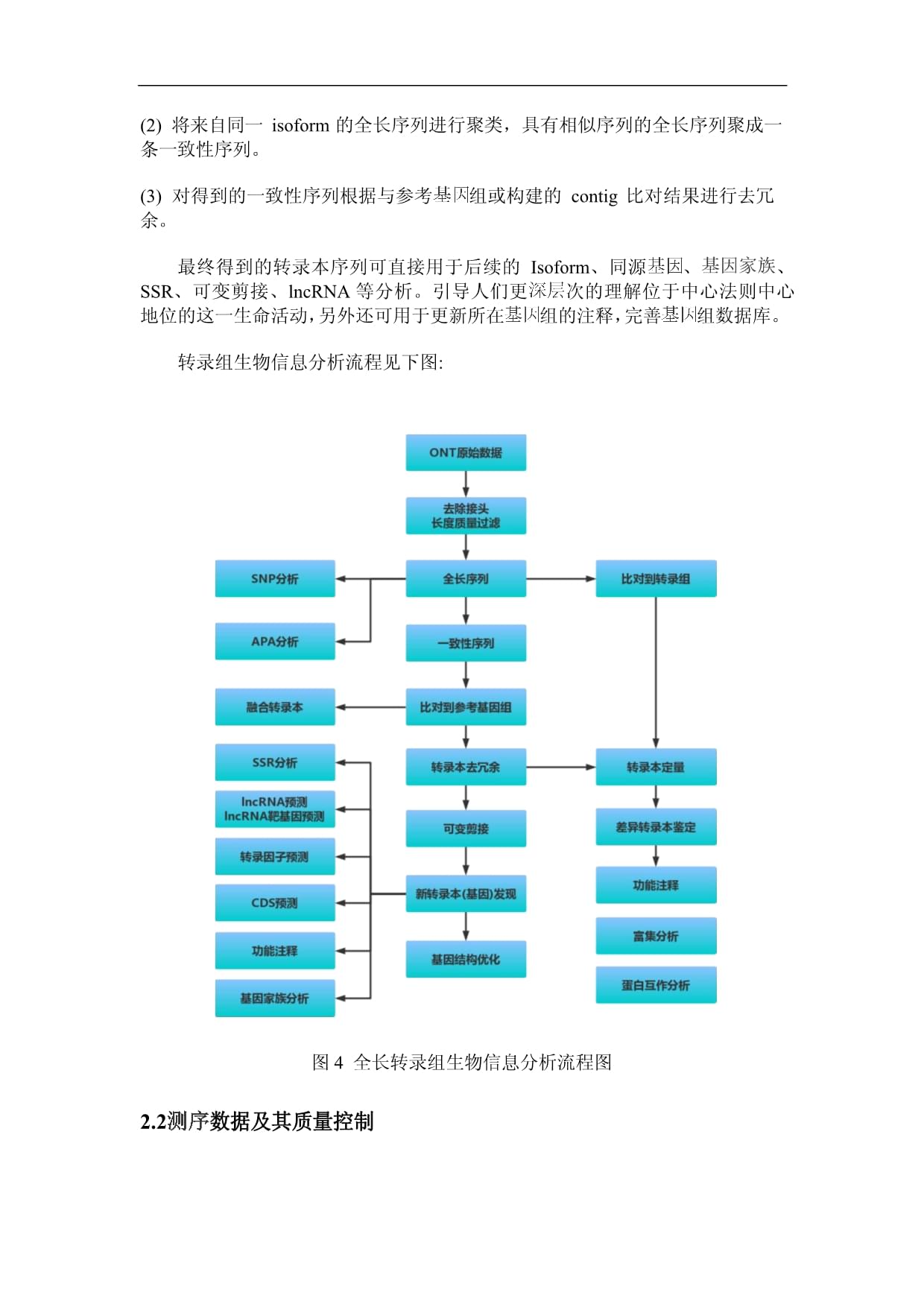
6個ONT有參全長轉錄組分合同關鍵指標完成6個樣品的全長轉錄組每個樣品產出不少于8GbCleanData。完成可變剪接分析。完成長鏈非編碼RNA預測和長鏈非編碼RNA靶預測。完成6個樣品的全長轉錄組共產出51.06Gbcleandata每個樣品的cleandata8Gb,每個樣品的全長(fulllength)257,235、257,235、257,235、257,235、257,235、257,235條。isoform)126,307、126,307、126,307、126,307、126,307、126,307個,一致性轉錄本序列通過GMAP比對到參考組后進行去冗余分析,最終分別獲得轉44,871、44,871、44,871、44,871、44,871、44,871條。475475475475475475共檢測到位點(geneloci)10,298個,其中新位點1,477個,新發現轉錄本32,154個。對新發現的轉錄本進行序列結構分析,本項目共預測出115,121個SSR、39,088ORF序列、39,0888,964個lncRNA62,4021.1原ONT是新一代基于納米孔的單分子實時電信號技術[1],其各平臺圖1ONT原目前Nanopore根據電流的大小及電流大小的變化情況,通過“遞歸神經網絡(RecurrentNeuralNetwork)”的復雜算法對堿基進行判讀[4]圖2ONT過程示例1.2實驗流OxfordNanoporeTechnologies(ONT)protocalRNANanodrop、Qubit0.35%瓊脂糖凝膠電泳進行純度、濃度引物退火,反轉錄成cDNAswitchRNADNA加上接頭,上機3Nanopore生物信息學分生物信息學分析流程概序平臺的Sq2.0技術往往不能準確得到或組裝出完整轉錄本,無法識別Ioform、同源、超、等位表達的轉錄本,使人們難以理解這一生命活動更次的含義。基于fordnoporehnologis單分子實時測序技術的全長轉錄組無須打斷N段,可反轉錄得到全長。該平臺的超長包含了單條完整轉錄本序列信息后期分析無需組裝所測即所得3個階段,全長序列識別、isoform水polyA,并根據序列中是否存在strandswitchingoligo序列和PolyA得到全長序列。isoform的全長序列進行聚類,具有相似序列的全長序列聚成一對得到的一致性序列根據與參考組或構建的contig比對結果進行去冗最終得到的轉錄本序列可直接用于后續的Isoform、同源、、SSR、可變剪接、lncRNA等分析。引導人們更次的理解位于中心法則中心地位的這一生命活動另外還可用于更新所在組的注釋完善組數據庫。轉錄組生物信息分析流程見下圖4數據產出統Nanopore的下機數據的原始數據格式為包含所有原始信號的二代fast5Readsfast5MinKNOW2.1軟件包中的Albacore軟件進行basecalling后會將fast5格式數據轉換為fastq原始fastq數據進一步過濾短片段和低質量的reads,去除接頭后,得到總的CleanData,其信息統計如下表所示。1CleanDataMeanLength:reads平均長度;MaxLengthReads長度;MeanQscore:平均質量值。reads平均質量值分布圖如下所示:5CleanData注:橫坐標表示reads平均質量值;縱坐標表示reads將reads10份,每一份統計reads6根據cDNA原理,reads兩端有switcholigo和ployT/A則判斷為全長轉全長轉錄本的reads平均質量值分布圖、長度分布圖以及長度與質量值之間78N509一致性轉錄本序列統得到的全長轉錄本用minmap2reads之間的信息,最后使用racon軟件得到一致性序列,一致性序列信息統計如下表所示。2一致性序列統計信息Sample注:SampleName:樣品名;SeqNum:序列條數;MeanLength:一致性序列平10N50轉錄本去冗冗余序列。同時,全長轉錄本過程中,3'端因存在polyA結構,可以確定3'端比較完整,而5'端序列可能存在降解,導致同一轉錄本的不同拷貝分到不同的cluster中,如下圖綠色圈中所示,5'端差異造成不同轉錄本,導致冗余序列的產通過GMAP(GenomicMapandAlignmentProgram)[5]將得到的一致序列與參考組進行比對(設置參數--cross-species--allow-close-indels0),對identity0.9,coverage0.855’端外顯子有差異的比對,最終每個樣品分別得到非冗余轉錄本序列44,871、44,871、44,871、44,871、44,871、44,871條。轉錄組Reads與參考組序列比對結果文件(通常為BAM格式)、GenomicsViewer)進行可視化瀏覽。IGV具有以下特點:、能在不同尺度下顯示單個或多個Reads在參考組上的位置,包括Reads在各個上的分布情況和在注釋的外顯子、內含子、剪接接合區間區、能在不同尺度下顯示不同區域的Reads豐度,以反映不同區域的轉錄水平能顯示及其剪接異構體的注釋信息能顯示其他注釋信息既可以從服務器端各種注釋信息,又可以從本地加載注釋信息12IGV結構優由于使用的軟件或數據本身的局限性導致所選參考組的注釋往往不夠轉錄本結構利用pare[6]將全長得到的轉錄本與組的已知轉錄本進行比較,發現新的和轉錄本,對組注釋進行補充。如果在原有邊界之外的區域有MappedReads支持,將的非翻譯區(UntranslatedRegion,UTR)向上下游延伸,修正的邊界。可變剪接分轉錄生成的前體mRNA(pre-mRNA),有多種剪接方式,選擇不同的外顯子,產生不同的成熟mRNA,從而翻譯為不同的蛋白質,構成生物性狀的通過Astalavista軟件[7]獲取每個樣品存在的可變剪接類型,主要的可圖13可變剪接類注:(A)外顯子跳躍;(B)可變轉錄終止位點;(C)可變外顯子;(D)(E)Astalavista軟件分析結果中,5種可變剪接事件情況進14::Exonskip:外顯子跳躍;Intronretention:內含子保留;Mutuallyexclusiveexon:可變外使用去冗余前的一致序列按下列條件對每個樣品進行融合轉錄本篩選2個或多個位點10kb475、475、475、475、475、475多聚腺苷酸化是指多聚腺苷酸與信使RNA(mRNA)分子的共價鏈結。在mRNA的方式的一部份。mRNA3'端中斷。多聚腺苷酸尾(或聚A尾)保護mRNA,免受核酸外切酶,并且對轉錄終結、將mRNA從細胞核輸出及進行翻譯都十分重要。前體mRNA的可變多聚腺苷酸化(alternativepolyadenylation,APA)可能貢獻于轉錄組多樣性,組的編碼能力以及的調控機制。我們采用TAPISpipeline[8]來識別APA。圖15多聚腺苷酸化位點個數分注:橫坐標:多聚腺苷酸化位點個數;縱坐標:個數利用MEME對所有轉錄本polyA50bp的motif如下圖所示:16polyAmotifSSR分MISA(MIcroSAliteidentificationtool)[9]是一款鑒定簡單重復序列的軟件,其參考見附表。它可以通過對轉錄本序列的分析,鑒定出7種類型的SSR:Mono-nucleotide(單堿基)、Di-nucleotide(雙堿基)、Tri-nucleotide(三(六堿基)、compoundSSR(混合微,兩個SSR距離小于100bp)500bpMISASSR分析,結3SSRSearchingTotalnumberofsequencesTotalsizeofexaminedsequences(bp)TotalnumberofidentifiedNumberofSSRcontainingNumberofsequencescontainingmorethan1SSRNumberofSSRspresentincompoundformationMonoDiTriTetraPentaHexa對不同SSR類型的密度分布進行統計,結果見下圖17SSR新編碼區序列預TransDecoder[10]v3.0.0)軟件基于開放閱讀框(OpenReadingFrame,ORF)長度、對數似然函數值(Log-likelihoodScore)Pfam數據庫蛋(CodingSequence,CDS),CDSTransDecoder軟件對其編碼區序列及其對應氨基酸orf63,879orf39,088CDS18CDSORF19CDS轉錄因子分RNADNA模板的結合,從而調控的轉錄。植物轉錄因子預測使用iTAK[11]軟件,動物轉錄因子鑒定使用動物轉錄因子數據庫——animalTFDB2.0[12],本項目對得到的新轉錄本共預測得4,968個。對不同類型的轉錄因子個數進行統計,結果見下圖:20LncRNA預因lncRNA不編碼蛋白,因此,通過對轉錄本進行編碼潛能篩選,判斷其是否具有編碼潛能,從而可以判定該轉錄本是否為lncRNA。分別應用CPC[13]分錄本進行lncRNAlncRNA8,964CPC分CPC(CodingPotentialCalculator)是一種基于序列比對的蛋白質編碼潛能計算工具。通過將轉錄本與已知蛋白數據庫比對,CPC根據轉錄本各個編碼框的生物學序列特征評估其編碼潛能。Score<0時,為noncodingRNA。CNCI分CNCI(Coding-Non-CodingIndex)分析是一種通過相鄰核苷酸三聯體特征區完整的轉錄本和反義轉錄本進行預測。CNCI工具提供兩種比對模式:ve(脊椎物種);pl(植物物種),本項目選擇ve參數。當score<0NoncodingCPAT分ORFORFFickettHexamer得分來判斷轉錄本編pfamPfam數據庫是最全面的蛋白結構域注釋的分類系統。蛋白質是由一個或多個結構域組成的,而每個特定結構域的蛋白序列具有一定保守性。Pfam將蛋白質的結構域分為不同的蛋白通過蛋白序列的比對建立了每個的氨基酸HMMpfam數據庫做hmmscanlncRNA。為直觀展示分析結果,將以上4種鑒定得到的noncding4種分析結果取交集,用于后續lncRNA214種篩選方法根據lncRNA在參考組注釋信息(gff)上的位置,對lncRNA進行分類22lncRNAlncRNA靶預對預測得到lncRNA序列進行靶預測基于lncRNA與其靶的作用2種預測方法:第一種,lncRNA調控其鄰近的表達,主要根據lncRNA與mRNA的位置關系預測,定義中每100kbp范圍內存在差異表達的lncRNA與差異表達的第二種,lncRNA與mRNA由于堿基互補配對而產生作用,主要利用靶預測工具對我們的LncRNA進行靶預測將得到的新轉錄本序列與NR[18]、Swissprot[19],GO[20]、COG[21]、NRNCBISwissprotPIR(ProteinInformationResource)PRF(ProteinResearchFoundation)PDB(ProteinDataBank)GenBank和RefSeq的CDS數據翻譯過來的蛋白質數據信息。該數據庫見附表。COG(ClustersofOrthologousGroups)數據庫是對產物進行同源分類的KOG(euKaryoticOrthologGroups)數據庫是針對真核生物,基于直系同源關系,結合進化關系將來自不同物種的同源分為不同的Ortholog簇。來自同一Ortholog的具有相同的功能,這樣就可以將功能注釋直接繼承給KOG簇的其他成員。個個結構域組成的,而每個特定結構域的蛋白序列具有一定保守性。Pfam將蛋白質的結構域分為不同的蛋白通過蛋白序列的比對建立了每個的氨基HMM統計模型。GO(GeneOntology)數據庫是一個國際標準化的功能分類體系提供了一套動態更新的標準詞匯表來全面描述生物體中和產物的功能屬性該(molecularfunction)(cellularcomponent)和生物學過程(biologicalprocess),各自描述了產物可能行使的TermTerm名,比如“cell”、“fibroblastgrowthfactorreceptorbinding”或者“signaltransduction”,同時有一個唯一的,形如KEGG(KyotoEncyclopediaofGenesandGenomes)數據庫是系統分析產(DISEASE)、序列(GENES)及組(GENOME)等。利用該數據庫有助于把及表達信息作為一個整體的網絡進行研究。4NewIsoform注:Annotateddatabases:數據庫名稱;NewIsoformNumber:各功能數據庫注釋到的SNP/InDel分SP(SingleleotidePolymorpims)是指在組上由單個核苷酸變異rds與參考基因組序列的opHt2比對結果使用TK軟件[24]識別樣品與參考組間的單堿基錯配,識別潛在的SP位點。進而可以分析這些SP位點是否影響了的表達水平或者蛋白產物的種類。InDel(insertion-deletion)是指相對于參考組樣本中發生的小片段的缺失,該缺失可能含一個或多個堿基。GATK也能夠檢測樣品的缺(InDel)。InDel變異一般比SNP變異少,同樣反映了樣品與參考組之間的差異,并且編碼區的InDel會引起移碼突變,導致功能上的變化。GATK35bp3SNP2.0選,最終獲得可靠的SNP位點。據變異位點在參考組上的位置以及參考組上的位置信息可以得到變異位點在組發生的區域(間區、區或CDS區等),以及變異產生的影響(同義非同義突變等)。由于轉錄完成之后,mRNA除了需要加帽、加Ploy(A)mRNA會經歷RNA編輯(RNAediting),從而會產生單堿基的替換、、缺失。RNA編輯能使同一產生序列多樣的堿基替換的RNA編輯結果是一樣的。因此,通過轉錄組數據識別出SNP不免會含有RNA編輯的產物。SNP位點統根據SNPSNP(Transition)和顛換(Transversion)兩種類型。根據SNP位點的等位(Allele)數目,可以將SNP位點分為純合型SNP位點(只有一個等位)和雜合型SNP位點(兩個或多個等位)。不同物種雜合型SNP所占的比例存在差異。SNP位點數目、轉換類型比例、顛換類型比例以及雜合型SNP位點比例進行統計,如下表:7SNP點總數;IntergenicSNP:間區SNP位點總數;Transition:轉換類型的SNP位點數目在SNP位點數目中所占的百分比;TransversionSNPSNP位點數目中所占的百分比;HeterozygositySNP位點數目在總SNP位點數目中所占的百SNP31SNP注:橫軸為SNP突變類型,縱軸為相應的SNPSNP密度分將每個的SNP位點數目除以的長度,得到每個的SNP位點密度值,統計所有的SNP位點密度值并做密度分布圖。的SNP32SNP注:橫軸為上平均每1000bp序列中分布的SNP數目,縱軸為數SNP/InDel注SNPEffSNP,InDel注釋,SNP,InDel33SNP注:縱軸為SNP34InDel使用全長轉錄組與組已知轉錄本作為參考進行序列比對及后續分minimp2[26]將比對到參考轉錄組的Reads表5全長數據與參考轉錄組比對結果統計TotalMappedReads數目及在全長序列中占的百分比。轉錄組數據飽和度檢合格的轉錄組文庫是轉錄組的必要條件為了評估數據是否充足并滿足后續分析,對得到的轉錄本數進行飽和度檢測。由于一個物種的數目是有限的,且轉錄具有時間和空間特異性,因此隨著量的增加,檢測到的MappedData對檢測到的不同表達情況的轉錄本數目飽和情況進行模擬,繪制曲23注:本圖為隨機抽取10%、20%、30%……90%的總體數據單獨進行定量分析的結Readsreads數的百分比,縱15%GeneRPT10K范圍的百分比。轉錄組可以模擬成一個隨機抽樣的過程為了讓片段數目能反映轉錄本表達水平,需要對樣品中的MappedReads的數目進行歸一化。采用RPT10K/RPG10K(ReadsPerTranscript/Geneper10,000reads)[27]作為衡量轉錄本或表達水平的指標,RPT10K計算公式如下:注:公式中,readsmappedtotranscriptreads數目;totalreadsalignedinsample表示比對到參考轉錄組的片段總數。(1)25RPT10KRPT10K的對RPT10K分布:26RPT10KRPT10K的對數值。該圖從表達還可以評估差異表達的可靠性和輔助異常樣品的篩查。將相關系數r(PearsonCorrelationCoefficient)作為生物學重復相關性的評估指標。r21,說明兩個重復樣品相關性越強。百邁客保證對同一條件的所有生物學重復樣品進行同人同批樣品提取建庫同Run同Lane。27為研究在不同實驗處理下的表達模式,首先對選取樣品的FPKMFPKMK-means聚類分析,同一類的在不同的實驗處理下具有相似的變化趨勢具有相似變化趨勢的往28注:x軸表示實驗分組,yFPKMcluster中的所有差異表達分低,差異表達可以劃分為上調(Up-regulatedGene)和下調A中的表達水之為下調。上調和下調是相對的,由所給A和B的順差異表達篩獲得兩個生物學條件之間的差異表達集;對于沒有生物學重復的樣本,使用在差異表達過程中將FoldChange≥2且FDR<0.01作為篩選標準。Rate)是通過對差異顯著性p值(p-value)進行校正得到的。由于轉錄組的差異表達分析是對大量的表達值進行獨立的統計假設檢驗會存在假陽性問Benjamini-Hochberg校正方p值(p-value)FDR作為差異表達篩選的關鍵指標。差異表 數目統計如下表表6差異表達數目統計DEGDEGup-down-注:DEGSet:差異表達集名稱;DEGNumber:差異表達數目;up-regulated:上調的數目;down-regulated:下調數目。通過火山圖(VolcanoPlot)可以快速地查看在兩個(組)樣品中表達圖29差異表達火山通過MA圖可以直觀地查看的兩個(組)樣品的表達水平和差異倍數MA圖見下圖:圖30差異表達MA差異表達聚類分對篩選出的差異表達做層次聚類分析將具有相同或相似表達模式的基因進行聚類,差異表達聚類結果如下圖:圖31差異表達聚類差異表達功能注釋和富集分對差異表達進行數據庫的功能注釋各差異表達集注釋到的數表7注釋的差異表達數量統計DEGSwiss-差異表達GO注釋及富GO數據庫是GO組織(GeneOntologyConsortium)2000年構建的一個差異表達GO分類統計結果見下圖圖32差異表達GO注釋分類統計差異表達KEGG注釋及富在生物體內,不同的產物相互協調來行使生物學功能,對差異表達的通路(Pathway)注釋分析有助于進一步解讀的功能。KEGG(KyotoEncyclopediaofGenesandGenomes)是系統分析功能、組信息數據庫,它有助于研究者把及表達信息作為一個整體網絡進行研究。作為是有關Pathway的主要公共數據庫(Kanehisa,2008)KEGG提供的整合代謝途徑包含有氨基酸序列、PDB庫的等等,是進行生物體內代謝分析、代謝網絡差異表達的通路注釋結果見下圖34KEGG對差異表達KEGG的注釋結果按照KEGG中通路類型進行分類,分類圖35差異表達KEGG分類注:縱坐標為KEGG代謝通路的名稱,橫坐標為注釋到該通路下的個數及其個數占被注釋上的總數的比例。差異表達的通路富集分析。Pathway顯著性富集分析以KEGG數據庫中Pathway為單位,應用超幾何檢驗,找出與整個背景相比,在差異表達中顯著性富集的Pathway。差異表達KEGG通路富集分析結果見下圖,圖中呈現了顯著性Q20個通路。圖36差異表達KEGG通路富集散點差異表達COG分COG(ClusterofOrthologousGroupsofproteins)數據庫是基于細菌、藻類、真核生物的系統進化關系構建得到的,利用COG數據庫可以對產物進行直差異表達COG分類統計結果見下圖圖37差異表達COG注釋分類統計差異表達eggNOG分eggNOG(evolutionarygenealogyofgenesnon-supervisedorthologousgroups)數據庫對直系同源類群進行了功能描述和功能分類的注釋,包含了1133個物種eggNOG分類統計結果見下圖:圖38差異表達eggNOG注釋分類統計差異表達蛋白互作網STIG[30]是收錄多個物種預測的和實驗驗證的蛋白質-蛋白質互作的數構建差異表達互作網絡對于數據庫中包含的物種可直接從數據庫中提取出目標集的互作關系對構建互作網絡對于數據庫中未收錄信息的物種使用T軟件將目的與數據庫中的蛋白質進行序列ytope軟件進行可視化。Cytoscape[31]可視化的差異表達蛋白質互作網絡如下圖39差異表達轉錄本分(E差異表達轉錄本集,使用_”的方式命名。根據兩(組)樣品之間表達水(prgulatdrrt(nrgulatdrnript(組B中的表達水平高于樣(組A中的表達水之為下調轉錄本上調和下調是相對的,由所給A和B的順序決定。差異表達篩EBSeq進行差異分析。在差異表達轉錄本檢測過程中,將FoldChange≥2且FDR<0.01作為篩選標準。差異倍數(FoldChange)表示兩樣品(組)間表達量的比值。FDR(FalseDiscoveryRate)p值(p-value)進行校正得到的。由于轉錄組的差異表達分析是對大量的轉錄本表達值進行獨立的統計假設檢驗會存在假陽性問題,因此在進行差異表達分析過程中,采用了公認的Benjamini-Hochberg校正方法對原有假設檢驗得到的顯著性p值(p-value)進行校正,并最終采用FDR作為差異表達轉錄本篩選的關鍵指標。8DETup-down-注:DETSet:差異表達轉錄本集名稱;DETNumber:差異表達轉錄本數目;up-regulated:通過火山圖(VolcanoPlot)可以快速地查看轉錄本在兩個(組)樣品中表40MA圖可以直觀地查看轉錄本的兩個(組)樣品的表達水平和差異倍MA圖見下圖:41差異表達轉錄本MA差異表達轉錄本聚類分429DET26注:DETSet:差異表達轉錄本集名稱;Total:注釋到的差異表達轉錄本數目;第三列到最差異表達轉錄本GO注釋GO數據庫是GO組織(GeneOntologyConsortium)2000年構建的一個GO43GO對樣品間差異轉錄本進行富集分析,富集到的Term做topGO有向無環圖。GO富集分析的結果圖形化展示,分支代表包含關系,從上至下所定義的功能描述范圍越來越具體。差異表達轉錄本的topGO有向無44topGO差異表達轉錄本KEGG在生物體內,不同的產物相互協調來行使生物學功能,對差異表達的通路(Pathway)注釋分析有助于進一步解讀的功能。KEGG(KyotoEncyclopediaofGenesandGenomes)是系統分析功能、組信息數據庫,它有助于研究者把及表達信息作為一個整體網絡進行研究。作為是有關Pathway的主要公共數據庫(Kanehisa,2008)KEGG提供的整合代謝途徑包含有氨基酸序列、PDB庫的等等,是進行生物體內代謝分析、代謝網絡45KEGGKEGGKEGG中通路類型進行分類,分46KEGG分析差異表達轉錄本在某一通是否發生顯著差異(over-presentation)即PathwayKEGG數據庫中Pathway為單位,應用超幾何檢驗,找出與整個轉錄本背景相比,在差異表達轉錄本中顯著性富集的PathwayKEGG通路富集分析結果見下圖,圖中呈現了顯著性Q20個通路。47KEGG差異表達轉錄本COG分COG(ClusterofOrthologousGroupsofproteins)數據庫是基于細菌、藻類、真核生物的系統進化關系構建得到的,利用COG數據庫可以對產物進行直差異表達轉錄本COG48COG差異表達轉錄本eggNOG分49eggNOG差異表達轉錄本蛋白互作網STRING是收錄多個物種預測的和實驗驗證的蛋白質-蛋白質互作的數據庫,錄信息的物種,使用BLAST軟件,將目的轉錄本與數據庫中的蛋白質進行序列白質互作網絡可導入Cytoscape軟件進行可視化。Cytoscape50參考文DeamerD,AkesonM,BrantonD:Threedecadesofnanoporesequencing.NatBiotechnol2016,34:518-524.MagiA,SemeraroR,MingrinoA,GiustiB,D'AurizioR:Nanoporesequencingdataysis:stateoftheart,applicationsandchallenges.BriefBioinform2017.JainM,OlsenHE,PatenB,AkesonM:TheOxfordNanoporeMinION:deliveryofnanoporesequencingtotomicscommunity.GenomeBiol2016,17:239.KrishnakumarR,SinhaA,BirdSW,Jaya nH,EdwardsHS,SchoenigerJS,PaKD,BrandaSS,BartsS:SystematicandstochasticinfluencesontheperformanceoftheMinIONnanoporesequenceracrossarangeofnucleotidebias.SciRep2018,8:3159.WuTD,WatanabeCK(2005)GMAP:agenomicmapandalignmentprogramformRNAandESTsequences.Bioinformatics21:1859–1875.PMID:15728110. pare:classify,merge,trackingandannotationofGFFfilesbycomparingtoareferenceannotationGFF.FoissacS,SammethM.ASTALAVISTA:dynamicandflexibleysisofalternativesplicingeventsincustomgenedatasets.NucleicAcidsResearch2007,35(WebServerAbdelghanySE,HamiltonM,JacobiJL,etal.Asurveyofthesorghumtranscriptomeusingsingle-moleculelongreads[J].NatureCommunications,2016,7:11706.ThielT,MichalekW,VarshneyR,GranerA.ExploitingESTdatabasesforthedevelopmentandcharacterizationofgene-derivedSSR-markersinbarley(HordeumvulgareL.).TheoreticalandAppliedGenetics.2003;106(3):411–22.TransDecoder:FindCodingRegionsWithinZhengY,JiaoC,SunH,RosliHG,PomboMA,ZhangP,BanfM,DaiX,MartinGB,GiovannoniJJ,ZhaoPX,RheeSY,FeiZ(2016)iTAK:aprogramforgenome-widepredictionandclassificationofplanttranscriptionfactors,transcriptionalregulators,andproteinkinases.MolecularPlant9:1667-1670.Hong-MeiZhang,TengLiu,Chun-JieLiu,ShuangyangSong,XiantongZhang,WeiLiu,HaiboJia,YuXue,andAn-YuanGuo.AnimalTFDB2.0:aresourceforexpression,predictionandfunctionalstudyofanimaltranscriptionfactors.Nucl.AcidsRes.(28January2015)43(D1):D76-D81.L.Kong,Y.Zhang,Z.Q.Ye,X.Q.Liu,S.Q.Zhao,L.Wei,andG.Gao.2007.CPC:assesstheprotein-codingpotentialoftranscriptsusingsequencefeaturesandsupportvectormachine.NucleicAcidsRes36:W345-349.LiangSun,HaitaoLuo,DechaoBu,GuoguangZhao,KuntaoYu,ChanghaiZhang,YuanningLiu,RunShengChenandYiZhao*Utilizingsequenceintrinsiccompositiontoclassifyprotein-codingandlongnon-codingtranscripts.NucleicAcidsResearch(2013),:10.1093/nar/gkt646.WangL,ParkHJ,DasariS,WangS,KocherJP,LiW.CPAT:Coding-PotentialAssessmentToolusinganalignment-freelogisticregressionmodel.NucleicAcidsRes.2013Apr1;41(6):e74.:10.1093/nar/gkt006.FinnRD,BatemanA,ClementsJ,etal.Pfam:theproteinfamiliesdatabase.NucleicAcidsResearch,2013:gkt1223.LiJ,MaW,ZengP,etal.LncTar:atoolforpredictingtheRNAsoflongnoncodingRNAs[J].BriefingsinBioinformatics,2015,16(5):806.DengYY,LiJQ,WuSF,ZhuYP,etal.IntegratedNRDatabaseinProteinAnnotationSystemandItsLocalization.ComputerEngineering2006.,32(5):71-74. R,BairochA,WuCH,BarkerWC,etal.UniProt:theUniversalProteinknowledgebase.NucleicAcidsResearch2004Jan1;32(Databaseissue):D115-9.AshburnerM,BallCA,BlakeJA,BotsteinD,etal.Geneontology:toolfortheunificationofbiology.NatureGenetics2000,25(1):25-29.TatusovRL,GalperinMY,NataleDA.TheCOGdatabase:atoolforgenomescaleysisofproteinfunctionsandevolution.NucleicAcidsResearch2000,28(1):33-36.KooninEV,FedorovaND,JacksonJD,etal.Acomprehensiveevolutionaryclassificationofproteinsencodedincompleteeukaryoticgenomes.Genomebiology,2004,5(2):R7.KanehisaM,GotoS,KawashimaS,OkunoY,etal.TheKEGGresourcefordecipheringtome.NucleicAcidsResearch2004,32(Databaseissue):D277-D280.MckennaA,HannaM,BanksE,etal.TheGenomeysisToolkit:aMapReduceframeworkforyzingnext-generationDNAsequencingdata[J].GenomeResearch,2010,20(9):1297-303.CingolaniP,PlattsA,LeLW,etal.Aprogramforannotatingandpredictingtheeffectsofsinglenucleotidepolymorphisms,SnpEff[J].Fly,2012,6(2):80.HengLi;Minimap2:pairwisealignmentfornucleotidesequences,Bioinformatics,Volume34,Issue18,15September2018,Pages3094–3100.ByrneA,BeaudinAE,OlsenHE,etal.Nanoporelong-readRNAseqrevealswidespreadtranscriptionalvariationamongthesurfacereceptorsofindividualBcells[J].NatureCommunications,2017.AndersS,HuberW.Differentialexpressionysisforsequencecountdata[J].GenomeBiology,2010,11(10):R106.LengN,DawsonJA,ThomsonJA,etal.EBSeq:anempiricalBayeshierarchicalmodelforinferenceinRNA-seqexperiments[J].Bioinformatics,2013,29(8):1035.FranceschiniA,SzklarczykD,FrankildS,etal.STRINGv9.1:protein-proteininteractionnetworks,withincreasedcoverageandintegration.[J].NucleicAcidsResearch,2013,41(Databaseissue):D808.ShannonP,MarkielA,OzierO,etal.Cytoscape:ASoftwareEnvironmentforIntegratedModelsofBiomolecularInteractionNetworks[J].GenomeResearch,2003,OxfordNanoporeTechnologiesLongReadRawreadswerefirstfilteredbyNanoFiltwithminimumaveragereadqualityscore=7andminimumreadlength=50bpandthentrimmedwithadaptersfromreadsendsandsplitedwithinternaladapters.Next,full-length,non-chemiric(FLNC)transcriptsweredeterminedbysearchingforthepolyA/Tsignalandstrandswitchingoligointrimmedreads.Overlapsbetweenthetrimmedreadsidentifiedbyminmap2with-xava-ontwereusedtoobtainconsensusisoformsbyRaconwithdefaultparameters.MoveConsensussequencesweremappedtoreferencegenomeusingGMAP.MappedreadswerefurthercollapsedbycDNA_Cupcakepackagewithmin-coverage=85%andmin-identity=90%.5’differencewasnotconsideredwhencollapsingredundanttranscripts.FindfusionThecriteriaforfusioncandidatesisthatasingletranscriptmustmapto2ormoreminimumcoverageforeachlociis5%andminimumcoverageinbpis>=1totalcoverageis>=distancebetweenthelociisatleast Transcriptswerevalidatedagainstknownreferencetranscriptannotationswith pare.ASeventsincludingIR,ES,AD,AAandMEEwereidentifiedbytheAStalavistatool.SSRofthetranscriptomewereidentifiedusingMISA.APAysiswasconductedwithTAPIS.CDSwerepredictedbyTransDecoder.TranscriptionfactorsPlanttranscriptionfactorswereidentifiedwithiTAK,whileAnimaltranscriptionfactorswereidentifiedfromanimalTFDB. Fourcomputationalapproachesinclude CI/CPAT/Pfam/werecombinedtosortnon-proteincodingRNAcandidatesfromputativeprotein-codingRNAsinthetranscripts.Putativeprotein-codingRNAswerefilteredoutusingaminimumlengthandexonnumberthreshold.Transcriptswithlengthsmorethan200ntandhavemorethantwoexonswereselectedaslncRNAcandidatesandfurtherscreenedusing CI/CPAT/Pfamthathavethepowertodistinguishtheprotein-codinggenesfromthenon-codinggenes.GenefunctionalGenefunctionwasannotatedbasedonthefollowingdatabases:NR(NCBInon-redundantproteinsequences);Pfam(ProteinKOG/COG/eggNOG(ClustersofOrthologousGroupsofSwiss-Prot(Amanuallyannotatedandreviewedproteinsequencedatabase);KEGG(KyotoEncyclopediaofGenesandGenomes);GO(Gene Picard-toolsv1.41andsamtoolsv0.1.18wereusedtosort,removeduplicatedreadsandmergethebamalignmentresultsofeachsample.GATK2orSamtoolssoftwarewasusedtoperformSNPcalling.RawvcffileswerefilteredwithGATKstandardfiltermethodandotherparameters(clusterWindowSize:10;MQ0>=4and(MQ0/(1.0*DP))>0.1;QUAL<10;QUAL<30.0orQD<5.0orHRun>5),andonlySNPswithdistance>5wereretained.ficationofgene/transcriptexpressionlevelsandDifferential Fulllengthreadsweremappedtothereferencetranscriptomesequence.Readswithmatchqualityabove5werefurtherusedtofy.Geneexpressionlevelswereestimatedbyr
溫馨提示
- 1. 本站所有資源如無特殊說明,都需要本地電腦安裝OFFICE2007和PDF閱讀器。圖紙軟件為CAD,CAXA,PROE,UG,SolidWorks等.壓縮文件請下載最新的WinRAR軟件解壓。
- 2. 本站的文檔不包含任何第三方提供的附件圖紙等,如果需要附件,請聯系上傳者。文件的所有權益歸上傳用戶所有。
- 3. 本站RAR壓縮包中若帶圖紙,網頁內容里面會有圖紙預覽,若沒有圖紙預覽就沒有圖紙。
- 4. 未經權益所有人同意不得將文件中的內容挪作商業或盈利用途。
- 5. 人人文庫網僅提供信息存儲空間,僅對用戶上傳內容的表現方式做保護處理,對用戶上傳分享的文檔內容本身不做任何修改或編輯,并不能對任何下載內容負責。
- 6. 下載文件中如有侵權或不適當內容,請與我們聯系,我們立即糾正。
- 7. 本站不保證下載資源的準確性、安全性和完整性, 同時也不承擔用戶因使用這些下載資源對自己和他人造成任何形式的傷害或損失。
最新文檔
- 2025石油化工產品購銷合同
- 2025年貸款擔保的合同樣本
- 縣汽車站建設招標合同
- 北海個人租房合同
- 農業產品采購合同
- 2025簡約住宅裝修合同范本
- 隔壁同意建房協議書
- 2025年03月如東縣事業單位工作人員120人筆試歷年典型考題(歷年真題考點)解題思路附帶答案詳解
- 2025年03月吉安縣敦城人力資源服務有限公司吉安縣政務服務大廳工作人員筆試歷年典型考題(歷年真題考點)解題思路附帶答案詳解
- 南寧理工學院《混凝土結構設計》2023-2024學年第二學期期末試卷
- (新教材)湘科版三年級下冊科學 1.2能溶解多少 教學課件
- ICP-AES分析原始記錄
- 《HSK標準教程2》第2課課件-(2)
- 【課件】3.2 DNA的結構課件 2021——2022學年高一下學期生物人教版必修2
- 第五章仿生原理與創新設計ppt課件
- 棗莊防備煤礦有限公司“7.6”重大火災事故詳細分析
- 口腔科診斷證明書模板
- 小學數學問題解決(吳正憲)
- 第五節 胡靜-常用正頜外科手術
- 礦井開拓方案比較
- DB23-黑龍江省建設工程施工操作技術規程-城鎮道路工程.doc
評論
0/150
提交評論