


版權說明:本文檔由用戶提供并上傳,收益歸屬內容提供方,若內容存在侵權,請進行舉報或認領
文檔簡介
1、一、中藥有效成分的提取1. 常用方法溶劑法浸漬法有效成分遇熱不穩定的或含大量淀粉、樹膠、果膠、黏液質的中藥的提取。出膏率低、以水為溶劑的時候提取液易發霉。煎煮法揮發性或對熱不穩定的藥物不適用回流提取法對熱不穩定的藥物不適用,溶劑用量大,操作麻煩連續回流提取法彌補了回流提取法溶劑用量大的不足。耗時長滲漉法溶劑用量大,費時長,操作麻煩水蒸氣蒸餾法揮發性、可隨水蒸氣蒸餾而不被破壞、難溶或不溶于水的化學成分。升華法具升華性的成分超臨界萃取法超聲波提取法不會改變成分的結構、縮短提取時間、提高提取效率。2. 溶劑法的要點溶劑提取法是根據中藥中各成分的溶解性,選擇適當的溶劑將中藥中的化學成分從藥材中提取出來
2、。 溶劑選擇的原則“相似相溶”原則溶劑的極性親水性越強,極性越大;親脂性越強,極性越小。溶劑按極性大小可分為親水性溶劑(極性較大)和親脂性溶劑(極性較小)。極性由小大:石油醚四氯化碳苯二氯甲烷氯仿乙醚乙酸乙酯正丁醇丙酮乙醇甲醇水水無機鹽、糖、氨基酸、蛋白質、有機酸鹽、生物堿鹽、苷類乙醇高濃度提取親脂性成分,低濃度提取親水性成分石油醚、苯、乙醚、氯仿、乙酸乙酯揮發油、油脂、葉綠素、樹脂、內酯、某些生物堿及一些苷元二、分離與精制1. 根據物質的溶解度差別進行分離1.1 利用溫度不同引起溶解度的改變進行分離結晶和重結晶溶劑不與重結晶物質發生化學反應對待結晶的成分熱時溶解度大,冷時溶解度小對雜質的溶解
3、度冷熱都易溶或冷熱都不溶溶劑的沸點較低,容易揮發,易與結晶分離除去無毒或毒性很小,便于操作單一溶劑:常用的溶劑有水、冰乙酸、甲醇、乙醇、丙酮、乙醚、氯仿、苯、四氯化碳、石油醚、二硫化碳等。混合溶劑:把對此物質溶解度很大和溶解度很小的兩種溶劑混合在一起,可以獲得良好的溶解性能。常用的混合溶劑有乙醇一水、乙醚一甲醇、乙酸一水、乙醚一丙酮等。純度判斷色譜(TLC或PC)三種展開系統中(Rf值0.2、0.5、0.8)單一斑點一定的晶型和均勻的色澤1.2 利用兩種以上不同溶劑的極性和溶解性差異穩定一致的熔點、熔距窄(1-2)1.3 利用酸堿進行分離HPLC或GC單峰1.4 利用沉淀試劑進行分離2. 利用
4、物質在兩相溶劑中的分配比不同進行分離2.1 液-液萃取法(兩相溶劑萃取法)利用混合物中各成分在兩種不相混溶的溶劑中分配系數的不同而達到分離的方法。分配系數K和分離因子,CU是溶質在上相溶劑中的濃度,CL是溶質在下相溶劑中的濃度下標A、B分別代表不同的兩種物質。(KAKB)100,僅作一次簡單萃取就可實現基本分離;100>l0,則需萃取10-12次;2時,要想實現基本分離,需作 100次以上萃取才能完成;1時,KAKB,意味著兩者性質相近,無法分離。分配比與pH酸性、堿性及兩性有機化合物,都具有游離型和解離型,二者可互相轉化,故在兩相中的分配比不同。pH3 時,酸性物質多呈非解離狀態(HA
5、)、堿性物質呈解離狀態(BH+)存在;PH12 時,則酸性物質呈解離狀態(A)、堿性物質呈非解離狀態(B)存在。可利用pH梯度萃取分離物質。2.2 液-液萃取與紙色譜一般50時,簡單萃取即可解決問題;但50時,則宜使用逆流分溶法。利用紙色譜可以選擇設計液-液萃取分離物質的最佳方案。2.3 液-液分配柱色譜分類固定相流動相分離物質正相色譜強極性溶劑,如水、緩沖溶液弱極性有機溶劑,氯仿、乙酸乙酯、丁醇水溶性或極性較大的成分如生物堿、苷類、糖類、有機酸等反相色譜石蠟油強極性溶劑,水或甲醇脂溶性化合物,如高級脂肪酸、油脂、游離甾體3. 根據物質的吸附性差別進行分離固-液吸附應用最多,分為:物理吸附()
6、表面吸附,無選擇性,可逆,進行快速,應用較廣。三要素:吸附劑、溶質、溶劑化學吸附有選擇性,吸附牢固,有時不可逆,應用較少。半化學吸附吸附力介于物理吸附和化學吸附之間。物理吸附的基本規律:極性相似者易于吸附。硅膠、氧化鋁(極性吸附劑)極性強者優先被吸附;在弱極性溶劑中吸附能力強。活性炭(非極性吸附劑)對非極性物質有較強親和能力;在水(極性溶劑)中吸附能力強。極性強弱的判斷:化合物的極性由分子中所含官能團的種類、數目及排列方式決定。吸附柱色譜用于物質的分離吸附劑用量一般為樣品量的30-60倍,通常100目;有干法上樣和濕法上樣兩種。盡可能選擇極性小的溶劑裝柱和溶解樣品。洗脫用溶劑的極性宜逐步增加,
7、跳躍不能太大。為避免化學吸附,酸性物質宜用硅膠,堿性物質宜用氧化鋁進行分離。通常在分離酸性(堿性)物質時,在溶劑中加入適量乙酸(氨、吡啶、二乙胺),防止拖尾、促進分離。溶劑系統可通過TLC進行篩選,使組分Rf值達到0.2-0.3的溶劑系統可以選用。聚酰胺色譜屬于氫鍵吸附(分子間氫鍵),特別適合分離酚類、醌類、黃酮類化合物。酰胺羰基與酚類、黃酮類化合物的酚羥基,或酰胺鍵上的游離氨基與醌類、脂肪羧酸上的羰基吸附強弱取決于各種化合物與之形成氫鍵締合的能力。形成氫鍵的基團數目越多,吸附能力越強。成鍵位置對吸附力也有影響。易形成分子內氫鍵者,其在聚酰胺上的吸附相對減弱。分子中芳香化程度高者,則吸附性增強
8、;反之則減弱。含水溶劑中的吸附規律溶劑的洗脫能力由弱強: 水甲醇氫氧化鈉水溶液甲酰胺二甲基甲酰胺(DMF)尿素水溶液對鞣質的吸附能力特別強,幾乎不可逆,故可用于植物粗提物的脫鞣質處理。大孔樹脂具有選擇性吸附(范德華力或氫鍵)和分子篩(樹脂本身的多孔性網狀結構)的性能。極性小的化合物在水中易被非極性樹脂吸附,極性化合物在水中易被極性樹脂吸附。洗脫劑極性越弱,洗脫能力越弱。一般先用蒸餾水洗,再用濃度逐漸增加的乙醇或甲醇洗脫。多糖、蛋白質、鞣質等水溶性雜質會隨水流出,極性小的物質后被洗出。廣泛應用與天然產物的分離和富集。4. 根據物質分子大小差別進行分離常用透析法、凝膠過濾法、超濾法和超速離心法。凝
9、膠過濾色譜也叫凝膠滲透色譜/分子篩過濾/排阻色譜。利用分子篩分離物質。可用于分離分子量1000以下的化合物。樣品中的大分子被排阻,先流出;小分子能滲透到凝膠顆粒內部,較晚流出。葡聚糖凝膠(Sepadex-G)只適于在水中應用;羥丙基葡聚糖凝膠(Sephadex-LH-20)既可在水中,又可在由極性與非極性溶劑組成的混合溶劑中應用。膜分離法主要包括滲透、反滲透、超濾、電滲析和液膜技術等。透析法多用于水溶性的大分子和小分子物質的分離,如蛋白質、酶、多糖分離過程中的脫鹽。按照孔徑大小,可將透析膜分為:微濾膜、超濾膜、反滲透膜、納米膜。5. 根據物質解離程度不同進行分離離子交換法:以離子交換樹脂作為固
10、定相,以水或含水溶劑作為流動相進行分離的一種方法離子交換:當流動相流過交換柱時,其中的中性物質及不與離子交換樹脂發生交換的離子將通過柱子流出,而可與樹脂上的離子交換基團發生交換的離子被吸附在樹脂上,隨后改變條件,并用適當的溶劑從柱上洗脫下來。陽離子交換樹脂:強酸型(磺酸根)和弱酸型(羧酸根)陰離子交換樹脂:強堿型和弱堿型用于:不同電荷離子的分離(中藥水提物中酸性、堿性及兩性化合物的分離);相同電荷離子的分離(酸堿性不同)。6. 根據物質沸點進行分離分餾法:利用中藥中各成分沸點的判別進行提取分離。適用于液體混合物的分離,如揮發油和一些液體生物堿的提取分離。三、中藥化學成分的結構研究方法質譜(MS
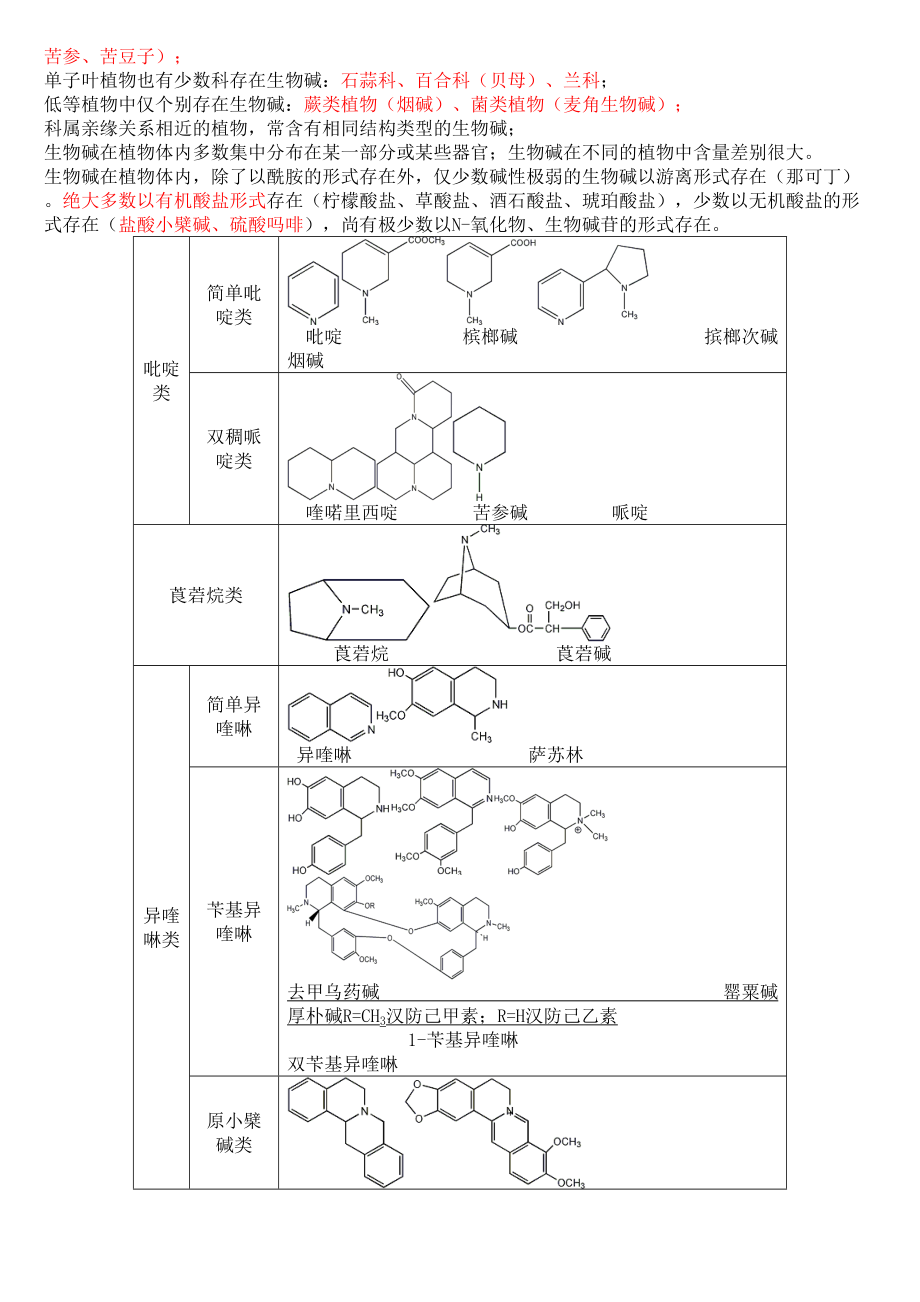
11、)電子轟擊質譜(EI-MS)需先將樣品加熱氣化后才能電離。不需加熱氣化的:化學電離(CI)、場致電離(FI)、場解析電離(FD)、快速原子轟擊電離(FAB)、電噴霧電離(ESI)紅外(IR)4000-1500cm-1區域為特征區域,可鑒別官能團;1500-600cm-1區域為指紋區,可鑒別化合物真偽。紫外-可見光譜UV光譜對共軛雙鍵、-不飽和羰基結構及芳香化合物的結構鑒定是重要手段1H-NMR通過化學位移、峰面積、信號的裂分及偶合常數(J)提供分子中質子的數目、類型及相鄰原子或原子團的信息。12C-NMR噪音去耦/全氫去耦/寬帶去耦:DEPT第二章 生物堿一、基本內容生物堿是指來源于生物界的一
12、類含氮有機化合物。大多有較復雜的環狀結構,氮原子結合在環內;多呈堿性,可與酸成鹽多具有顯著的生理活性。(例外:秋水仙堿,N原子不在環內,且幾乎不顯堿性)。絕大多數存在于雙子葉植物中:毛茛科(黃連、烏頭、附子)、防己科(漢防己、北豆根)、罌粟科(罌粟、延胡索)、茄科(洋金花、顛茄、莨菪)、馬錢科(馬錢子)、小檗科(三顆針)、豆科(苦參、苦豆子);單子葉植物也有少數科存在生物堿:石蒜科、百合科(貝母)、蘭科;低等植物中僅個別存在生物堿:蕨類植物(煙堿)、菌類植物(麥角生物堿);科屬親緣關系相近的植物,常含有相同結構類型的生物堿;生物堿在植物體內多數集中分布在某一部分或某些器官;生物堿在不同的植物中
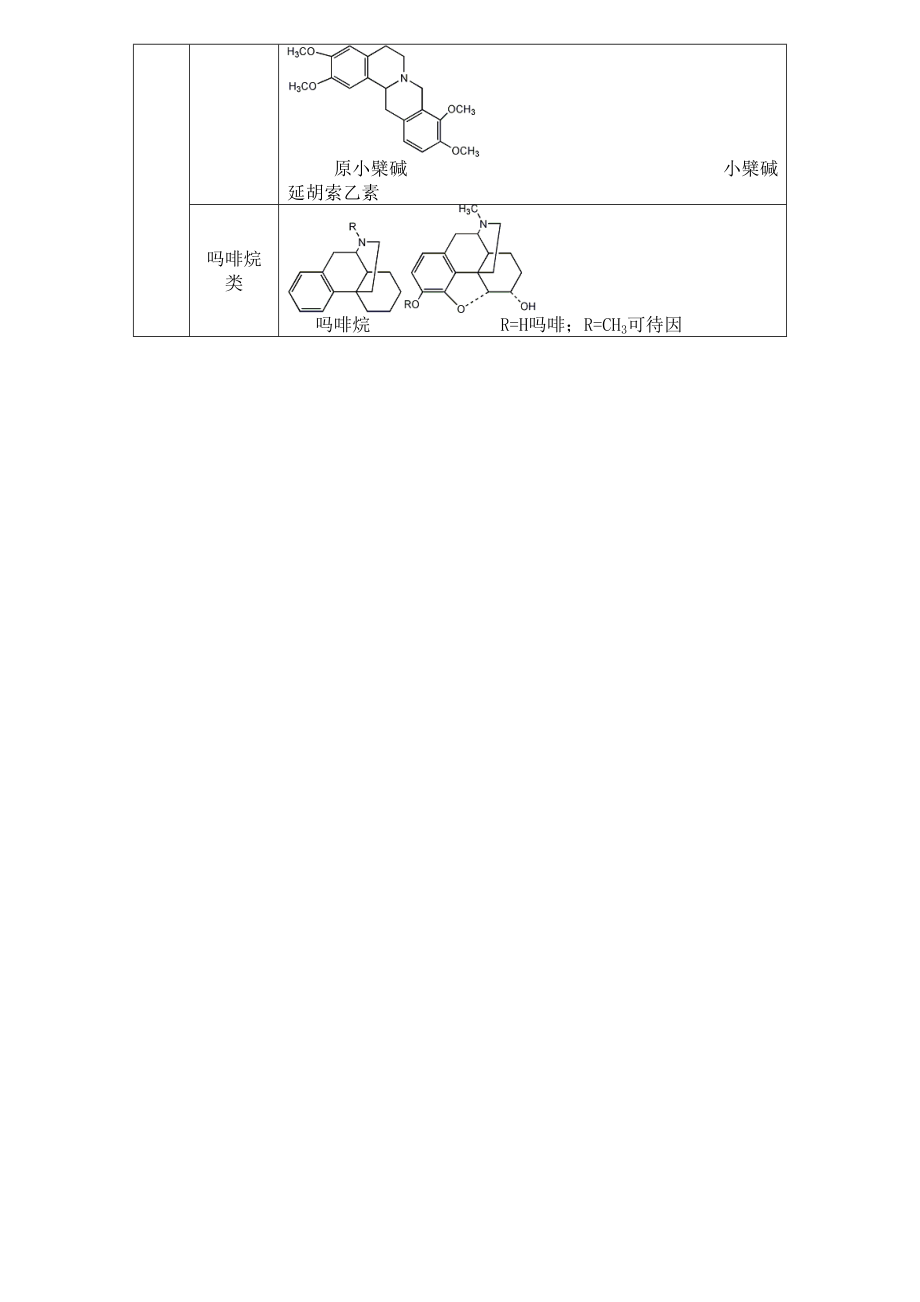
13、含量差別很大。生物堿在植物體內,除了以酰胺的形式存在外,僅少數堿性極弱的生物堿以游離形式存在(那可丁)。絕大多數以有機酸鹽形式存在(檸檬酸鹽、草酸鹽、酒石酸鹽、琥珀酸鹽),少數以無機酸鹽的形式存在(鹽酸小檗堿、硫酸嗎啡),尚有極少數以N-氧化物、生物堿苷的形式存在。吡啶類簡單吡啶類 吡啶 檳榔堿 擯榔次堿 煙堿雙稠哌啶類 喹喏里西啶 苦參堿 哌啶莨菪烷類 莨菪烷 莨菪堿異喹啉類簡單異喹啉 異喹啉 薩蘇林芐基異喹啉去甲烏藥堿 罌粟堿 厚樸堿R=CH3漢防己甲素;R=H漢防己乙素 1-芐基異喹啉 雙芐基異喹啉原小檗堿類 原小檗堿 小檗堿 延胡索乙素嗎啡烷類 嗎啡烷 R=H嗎啡;R=CH3可待因吲哚
14、類簡單吲哚類(吲哚)、大青素B、靛青苷單萜吲哚類(士的寧)、利血平色胺吲哚類(色胺) 吳茱萸堿雙吲哚類長春堿、長春新堿有機胺類(麻黃堿)、秋水仙堿、益母草堿二、理化性質性狀形態多數生物堿呈結晶形固體,有些為晶形粉末狀;少數生物堿為液體狀態(煙堿、毒芹堿、檳榔堿),分子中多無氧原子,或氧原子結合為酯鍵;個別具有揮發性(麻黃堿)、升華性(咖啡因、川芎嗪)。味道大多數生物堿具苦味;少數生物堿具有其他味道,如甜菜堿具有甜味。顏色絕大多數生物堿無色或白色;少數具有較長共軛體系和助色團的有一定顏色。如小檗堿、蛇根堿為黃色,藥根堿、小檗紅堿為紅色;有的生物堿在可見光下無色,在紫外光下顯熒光(利血平)。旋光性
15、含有手性碳原子或本身為手性分子的生物堿都有旋光性,且多呈左旋光性。生物堿的旋光性受手性碳的構型、測定溶劑、pH 值、溫度及濃度等的影響。麻黃堿在水中呈右旋性,在三氯甲烷中則呈左旋性;煙堿在中性條件下呈左旋性,在酸性條件下呈右旋性。溶解性游離生物堿親脂性生物堿多數具有仲胺和叔胺氮原子的生物堿,有較強的脂溶性。親水性生物堿季銨型生物堿:離子型,易溶于水和酸水。小分子生物堿:分子小而堿性強的,即可溶于水,也可溶于氯仿。麻黃堿、煙堿含N-氧結構的生物堿:含配位鍵結構,可溶于水。氧化苦參堿。酰胺類生物堿:可在水中形成氫鍵。秋水仙堿、咖啡堿具有特殊官能團的生物堿具酚羥基或羧基:兩性生物堿,可溶于酸水和堿水
16、。具酚羥基者可溶于氫氧化鈉等強堿性溶液,如嗎啡;具羧基者可溶于碳酸氫鈉溶液,如檳榔次堿。具內酯或內酰胺結構:正常情況下,溶解性類似一般叔胺堿,但在強堿溶液中加熱,可開環形成鹽而溶于水。如喜樹堿、苦參堿、藥跟堿、青藤堿、嗎啡等。例外嗎啡:酚性生物堿,但難溶于氯仿、乙醚;石蒜堿:難溶于有機溶劑,溶于水;喜樹堿:不溶于一般有機溶劑,溶于酸性氯仿。生物堿鹽一般易溶于水,可溶于甲醇、乙醇,難溶或不溶于親脂性有機溶劑。生物堿在酸水中成鹽溶解,調堿性后又游離析出沉淀,可利用此性質提取分離生物堿。生物堿鹽的水溶性因其成鹽的種類不同而有差異:生物堿的無機酸鹽有機酸鹽;無機酸鹽中含氧酸鹽鹵代酸鹽;小分子有機酸鹽大
17、大分子有機酸鹽。某些生物堿鹽難溶于水,如小檗堿鹽酸鹽、麻黃堿草酸鹽等難溶于水。生物堿的堿性:堿性強弱的表示方法:強堿pKa11季銨堿、胍類生物堿用其共軛酸的酸式離解指數pKa值表示。pKa值大,堿性強;pKa值小,堿性弱。中強堿pKa711脂胺、脂雜環類弱堿pKa27芳香胺、N-六元芳雜環類極弱堿pKa2酰胺、N-五元芳雜環類氮原子的雜化方式堿性隨軌道中s成分比例的增加而減弱,即sp3sp2sp脂胺類、脂氮雜環(sp3,中強堿);芳香胺類、六元芳雜環類(sp2,弱堿);氰基(sp,中性)電性效應增大氮原子未共用電子云密度,則堿性增強,反之則堿性減弱。誘導效應供電子基(烷基)使氮原子上電子云密度
18、增加,堿性增強;(麻黃堿去甲基麻黃堿)吸電子基(含氧基團、雙鍵、苯基)使氮原子上電子云密度減少,堿性減弱。共軛效應苯胺型:堿性減弱,如毒扁豆堿。酰胺型:堿性極弱,強胡椒堿、秋水仙堿、咖啡堿等空間效應氮原子附近取代基存在空間立體障礙,不利于接受質子,則堿性減弱。堿性:甲基麻黃堿麻黃堿;東莨菪堿山莨菪堿莨菪堿。氫鍵效應氮原子附近存在羥基、羰基等取代基團,并處于有利于形成穩定的分子內氫鍵時,其共軛酸穩定,堿性強。堿性:偽麻黃堿麻黃堿沉淀反應和顯色反應常用沉淀試劑碘化鉍鉀黃色至橘黃色無定形沉淀硅鎢酸淡黃色或灰白色無定形沉淀碘化汞鉀類白色沉淀飽和苦味酸黃色沉淀或結晶碘-碘化鉀紅棕色無定形沉淀雷氏銨鹽紅色
19、沉淀或結晶一般在酸性水溶液中進行,苦味酸試劑可在中性條件下進行。陽性判斷:一般需采用3種以上試劑分別進行反應。麻黃、嗎啡、咖啡堿等不與生物堿沉淀試劑反應,需用其它檢識方法。蛋白質、多肽、氨基酸、鞣質等非生物堿成分,也能與生物堿沉淀試劑作用產生沉淀。為避免干擾,可將酸水液堿化后用氯仿萃取出游離的生物堿,再用酸水自氯仿中萃取出生物堿。Mandelin試劑(1礬酸銨的濃硫酸溶液)莨菪堿及阿托品顯紅色,士的寧顯紫色,奎寧顯淡橙色。Frohde試劑(1鉬酸鈉或鉬酸銨的濃硫酸溶液)烏頭堿顯黃棕色,小檗堿顯棕綠色。Marquis試劑(含少量甲醛的濃硫酸)嗎啡顯紫紅色,可待因顯藍。三、生物堿的提取特殊提取方法
20、:水蒸氣蒸餾法(麻黃堿);升華法(咖啡堿、煙堿)1. 水或酸水提取常用0.1%-1%的硫酸或鹽酸溶液作為提取溶劑,采用浸漬或滲漉法提取。常采用陽離子交換樹脂、萃取法進一步純化和富集,除去水溶性雜質。2. 醇類溶劑提取游離的生物堿和生物堿鹽均可溶于甲醇、乙醇。常采用浸漬、滲漉、回流、連續回流法提取。3. 親脂性有機溶劑提取游離生物堿易溶于親脂性有機溶劑的性質,用氯仿、苯、乙醚以及二氯甲烷等溶劑。用親脂性有機溶劑提取之前,必須將中藥用堿水(石灰乳、碳酸鈉溶液或稀氨水)濕潤,使生物堿游離,再用親脂性有機溶劑萃取。提取方法多采用浸漬法、回流提取法或連續回流提取法。四、生物堿的分離1. 生物堿初步分離將
21、總生物堿按堿性強弱、酚性有無及是否水溶性,初步分離為5個部分。2. 生物堿單體的分離利用生物堿的堿性差異(pH梯度萃取)將總堿溶于稀酸水中,逐步加堿液調節pH,使pH由低到高,每調節一次pH,用氯仿萃取數次,從而將堿性由弱到強的生物堿依次轉溶于氯仿而得以分離。將總生物堿溶于氯仿中,用pH由高到低(8-3)的酸性緩沖液依次萃取,使生物堿按堿性由強至弱的順序自總堿中逐一轉溶至酸性緩沖液中;然后分別將各部分酸性緩沖液堿化,用氯傷萃取得到不同堿性的生物堿。利用生物堿及其鹽的溶解度差異苦參堿和氧化苦參堿:苦參堿的極性小于氧化苦參堿,前后能溶于乙醚,而后者難溶于乙醚漢防己甲素和漢防己乙素:漢防己乙素難溶于
22、苯,而漢防己甲素可溶于冷苯。麻黃堿和偽麻黃堿:前者的草酸鹽較后者的草酸鹽在水中的溶解度小。將麻黃堿和偽麻黃堿溶于適量水中,加入一定量草酸,麻黃堿生成草酸鹽即先從水溶液中析出。利用生物堿特殊官能團常見的有酚羥基、羧基、內酯或內酰胺結構等官能團。可利用這些官能團進行分離。色譜法吸附柱色譜:常用氧化鋁和硅膠作吸附劑,以親脂性有機溶劑為洗脫劑。分配柱色譜:以硅膠為支持劑,酸性緩沖液為固定相。五、色譜檢識TLC吸附薄層吸附劑:硅膠和氧化鋁,適用于脂溶性生物堿,氧化鋁的吸附力較硅膠強,更適合。展開劑:以氯仿為基本溶劑,適當調整極性,并常加入堿性溶劑(二乙胺、氨水等)防止拖尾:鋪堿板(0.1-0.5mol/
23、L的NaOH或緩沖液);用堿性展開劑;堿性環境(氨水)下展開。分配薄層硅膠或纖維素作為支持劑;甲酰胺或水作為固定相展開劑:脂溶性生物堿用親脂性有機溶劑(氯仿-甲苯);水溶性生物堿用親水性溶劑(BAW系統)以甲酰胺為固定相的薄層色譜,適于分離弱極性或中等極性的生物堿;以水為固定相的薄層色譜,適于分離水溶性的生物堿。PC多為正相分配色譜,常用于水溶性生物堿、生物堿鹽和弱親脂性生物堿的分離檢識。固定相: 水; 甲酰胺; 酸性緩沖液。展開劑:以水作固定相的紙色譜,宜用親水性溶劑系統作展開劑,如BAW【正丁醇-乙酸-水(4:1:5),上層】;以甲酰胺和酸性緩沖液作固定相的紙色譜,多以親脂性有機溶劑系統作
24、展開劑。HPLCGC具揮發性的生物堿:麻黃堿、煙堿六、含生物堿中藥實例1. 苦參化學結構苦參堿、氧化苦參堿(雙稠哌啶類)分子中均有2個氮原子,一個是叔胺氮,一個是酰胺氮。指標成分苦參堿、氧化苦參堿堿性叔胺氮(N-1),呈堿性;酰胺氮(N-16),幾乎不顯堿性,只相當于一元堿。溶解性苦參堿既可溶于水,又能溶于氯仿、乙醚、苯、二硫化碳親脂性溶劑;氧化苦參堿親水性比苦參堿更強,易溶于水,可溶于氯仿,難溶于乙醚。苦參堿的極性大小順序:氧化苦參堿羥基苦參堿苦參堿生物活性消腫利尿、抗腫瘤、抗病原體、抗心律失常、正性肌力、抗缺氧、擴張血管、降血脂、抗柯薩奇病毒、調節免疫2. 麻黃化學結構麻黃堿、偽麻黃堿,甲
25、基麻黃堿、甲基偽麻黃堿和去甲基麻黃堿、去甲基偽麻黃堿。(有機胺類)指標成分鹽酸麻黃堿性狀麻黃堿和偽麻黃堿的分子量較小,為無色結晶。兩者皆具有揮發性。堿性麻黃堿和偽麻黃堿為仲胺生物堿,堿性較強。偽麻黃堿的堿性稍強于麻黃堿。溶解性水溶性:游離的麻黃堿可溶于水,但偽麻黃堿在水中的溶解度較麻黃堿小。草酸麻黃堿難溶于水,而草酸偽麻黃堿易溶于水;鹽酸麻黃堿不溶于氯仿,而鹽酸偽麻黃堿可溶于氯仿。鑒別反應二硫化碳-硫酸銅反應產生棕色沉淀。銅絡鹽反應麻黃堿和偽麻黃的水溶液加硫酸銅、氫氧化鈉,溶液呈藍紫色。提取分離溶劑法;水蒸氣蒸餾法;離子交換樹脂法(利用強酸型陽離子交換樹脂,麻黃堿的堿性較偽麻黃堿弱,先從樹脂柱
26、上洗脫。)生物活性麻黃堿有收縮血管、興奮中樞神經的作用,能興奮大腦、中腦、延腦和呼吸循環中樞,有類似腎上腺素樣作用,能增加汗腺及唾液腺分泌,緩解平滑肌痙攣。偽麻黃堿有升壓、利尿作用。偽麻黃堿 (偽麻黃堿的共軛酸分子內氫鍵穩定) 麻黃堿化學結構小檗堿(黃連素)、巴馬丁、黃連堿、甲基黃連堿、藥根堿。均為季銨型生物堿。芐基異喹啉衍生物,屬于原小檗堿型。指標成分鹽酸小檗堿性狀自水或稀乙醇中析出的小檗堿為黃色針狀結晶。于160分解。鹽酸小檗堿為黃色小針狀結晶,加熱至 220左右分解,生成紅棕色小檗紅堿。小檗堿及其鹽類干燥時溫度不宜過高,一般不超過80。溶解性游離小檗堿能緩緩溶解于水中,易溶于熱水或熱乙醇
27、,在冷乙醇中溶解度不大,難溶于苯、氯仿、丙酮。小檗堿的鹽酸鹽在水中的溶解度較小,較易溶于沸水,難溶于乙醇。小檗堿與大分子有機酸(甘草酸、黃芩苷、大黃鞣質)結合的鹽在水中的溶解度都很小。配伍注意。小檗堿一般以季銨型生物堿的狀態存在,可以離子化呈強堿性,能溶于水,溶液為紅色。但在其水溶液中加入過量堿,季銨型小檗堿則部分轉變為醛式或醇式,其溶液也轉變成棕色和黃色。醇式和醛式小檗堿為親脂性成分,可溶于乙醚等親脂性有機溶劑。鑒別丙酮加成反應黃色結晶性小檗堿丙酮加成物。漂白粉顯色反應水溶液由黃色轉變為櫻紅色。生物活性小檗堿具有抗菌、抗病毒、抗炎作用。4. 川烏化學結構雙酯型生物堿烏頭堿、次烏頭堿和美沙烏頭
28、堿,為二萜生物堿,屬于四環或五環二萜類衍生物。指標成分烏頭堿、次烏頭堿、新烏頭堿主要毒性烏頭堿、次烏頭堿和美沙烏頭堿等雙酯型生物堿,毒性極強,是烏頭的主 要毒性成分炮制解毒將雙酯型堿經水解除去酯基,生成單酯型生物堿(烏頭次堿)或醇胺型生物堿(烏頭原堿),則毒性降低。5. 洋金花化學結構莨菪烷衍生物,主要包括莨菪堿、東莨菪堿、山莨菪堿、樟柳堿和N-去甲莨菪堿。其中阿托品是莨菪堿的外消旋體。指標成分硫酸阿托品、氫溴酸東莨菪堿堿性東莨菪堿和樟柳堿山莨菪堿莨菪堿鑒別氯化汞沉淀反應莨菪堿(或阿托品)加熱后沉淀變為紅色。東莨菪堿則與氯化汞反應生成白色沉淀。Vitali反應莨菪堿(或阿托品)、東莨菪堿等莨菪
29、烷類生物堿,顯深紫色。樟柳堿為負反應。過碘酸氧化乙酰丙酮縮合反應樟柳堿可發生該反應顯黃色,其余不反應。毒性中毒機制主要是M-膽堿反應。6. 馬錢子化學結構士的寧(番木鱉堿)和馬錢子堿,有強毒性,屬于吲哚類衍生物。鑒別硝酸反應士的寧與硝酸作用呈淡黃色,蒸干后的殘渣遇氨氣即變為紫紅色;馬錢子堿與濃硝酸接觸呈深紅色,繼加氯化亞錫,由紅色轉為紫色。濃硫酸-重鉻酸鉀反應士的寧初顯藍紫色,漸變為紫堇色、紫紅色,最后為橙黃色。馬錢子堿在此條件下不能產生相似的顯色反應。第三章 糖和苷一、糖的分類單糖是多羥基醛或酮,是組成糖類及其衍生物的基本多元。習慣上將單糖Fischer投影式中距羰基最遠的不對稱碳原子的構型
30、定為整個糖分子的絕對構型,其羥基向右的為D-型,向左的為L-型。根據成環的C原子多少,可分為五碳糖(呋喃糖)、六碳糖(吡喃糖)。單糖成環后新形成的一個不對稱碳原子成為端基碳,生成的一對差向異構體有、兩種構型。五碳醛糖D-木糖(xyl) D-核糖(rib)L-阿拉伯糖(ara)六碳醛糖D-半乳糖(gal) D-甘露糖(man) D-葡萄糖(glc)甲基五碳醛糖D-夫糖(fuc) L-鼠李糖(rha) D-雞納糖(qui)六碳酮糖D-果糖(fru)糖醛酸 D-葡萄糖醛酸 D-半乳糖醛酸由29分子個單糖通過苷鍵結合而成的直鏈或支鏈聚糖稱為低聚糖,或寡糖。具有游離醛基或酮基的糖稱為還原糖(槐糖、櫻草糖
31、);兩個單糖均以半縮醛或半縮酮上的羥基縮合成的聚糖為非還原糖(海藻糖、蔗糖)由10個以上單糖通過苷鍵連接而成的糖稱為多聚糖,或多糖。分兩類:一類是動植物的支持組織,該類成分不溶于水,分子呈直鏈型,如纖維素;一類是動植物的貯存養料,可溶于熱水成膠體溶液,多數分子呈支鏈型,如淀粉。淀粉由直鏈的糖淀粉和支鏈的膠淀粉組成。糖淀粉遇碘顯藍色,膠淀粉遇碘顯紫紅色。二、苷的分類按苷元的化學結構:氰苷、香豆素苷、木脂素苷、黃酮苷、蒽醌苷、吲哚苷等。按苷類在植物體內的存在狀況:原生苷、次生苷。苦杏仁苷是原生苷,水解后失去一分子葡萄糖而成的野櫻苷就是次生苷。按苷鍵原子O-苷醇苷:紅景天苷、毛莨苷、獐芽菜苦苷。強心
32、苷、皂苷酚苷:苯酚苷、萘酚苷、蒽醌苷、香豆素苷、黃酮苷、木脂素苷等。如天麻苷。氰苷:主要是指-羥腈的苷,苦杏仁苷。酯苷:既有縮醛性質又有脂的性質,易為稀酸和稀堿所水解。山慈菇苷A吲哚苷:靛苷S-苷蘿卜苷和芥子苷N-苷巴豆苷C-苷是一類不通過O原子,而直接以C原子與苷元的C原子相連的苷類。牡荊素、蘆薈苷。其他分類方法按苷的特殊性質分類,如皂苷;按生理作用分類,如強心苷;按糖的名稱分類,如木糖苷、葡萄糖苷;按連接單糖基的數目分類,如單糖苷、雙糖苷、叁糖苷;按連接的糖鏈數目分類,如單糖鏈苷、雙糖鏈苷等。三、化學性質氧化反應單糖分子中有醛(酮)基、伯醇基、仲醇基和鄰二醇基結構單元。通常醛(酮)基最易被
33、氧化,伯醇次之。Fehling反應在堿性試劑下,Ag及Cu2可將醛基氧化成羧基,分別生成金屬銀及磚紅色的氧化亞銅。溴水氧化糖的醛基生成糖酸。只氧化醛糖不氧化酮糖。過碘酸氧化反應在水溶液中進行。多用于糖苷類和多元醇的結構研究。可以推測出糖的種類、糖與糖的連接位置、分子中鄰二醇羥基的數目以及碳的構型等。羥基反應在糖和苷的羥基中,最活潑的是半縮醛羥基,次之是伯醇羥基,再次是C2-羥基包括:醚化、酰化、縮醛和縮酮化、硼酸絡合反應羰基反應糖的羰基還可被催化氫化或金屬氫化物還原,其產物叫糖醇。具有醛或酮羰基的單糖可與苯肼反應,首先生成腙,在過量的苯肼存在下繼續作用生產脎。四、苷的水解酸催化水解具有縮醛結構
34、,易為稀酸催化水解。一般在水或稀醇溶液中進行。常用的酸有鹽酸、硫酸、乙酸、甲酸。機制是苷原子先質子化,然后斷鍵生成碳正離子或半椅型中間體,在水中溶劑化而成糖。按苷鍵原子不同,酸水解的易難順序為:N-苷O-苷S-苷C-苷。吡喃糖苷中吡喃環的C-5上取代基越大越難水解。水解的易難順序為五碳糖>甲基五碳糖>六碳糖>七碳糖。如果接有-COOH,則最難水解。氨基糖較羥基糖難水解,羥基糖又較去氧糖難水解。呋喃糖苷較吡喃糖苷易于水解,酮糖較醛糖易水解。芳香屬苷因苷元部分有供電子基,水解比脂肪屬苷容易得多。苷元為小基團者,苷鍵橫鍵比苷鍵豎鍵的易于水解,因為橫鍵上原子易于質子化。堿催化水解僅酯
35、苷、酚苷、烯醇苷和-吸電子基取代的苷等才能被水解。 水楊苷酶催化水解專屬性高,條件溫和。可獲知苷鍵的構型,保持苷元結構不變,還可保留部分苷鍵得到次生苷或低聚糖。-果糖苷水解酶(轉化糖酶);-葡萄糖苷水解酶(麥芽糖酶);-葡萄糖苷水解酶(杏仁苷酶,水解一般-葡萄糖苷和有關六碳醛糖苷、纖維素酶)pH對酶水解十分重要(芥子苷酶水解芥子苷,在pH 7時酶解生成異硫氰酸酯,在pH 3-4時酶解則生成腈和硫黃)五、顯色:Molish反應:由濃硫酸和和-萘酚組成。可檢識糖和苷的存在。六、提取分離:一般采用水或醇抽提。提取苷類時,需要抑制酶的活性:采用甲醇、乙醇或沸水提取,或者在藥材原料中拌入一定量的無機鹽(
36、如碳酸鈣)。其次是在提取過程中要注意避免與酸或堿接觸,防止苷類水解。七、結構鑒定PC展開系統:以水飽和的有機溶劑。BAW、水飽和的苯酚。(增加Rf值可加入乙酸、吡啶、乙醇增加含水量)紙色譜檢識時,鼠李糖的Rf值一般大于葡萄糖顯色劑:硝酸銀試劑、三苯四氮唑鹽試劑、苯胺-鄰苯二甲酸鹽試劑、3,5-二羥基甲苯-鹽酸試劑。TLC點樣量不宜過多。若硅膠用0.03molL硼酸溶液或一些無機鹽的水溶液代替水調制吸附劑涂鋪薄層,則樣品承載量可明顯增加,分離效果也有改善。分子量的測定MS。一般采用場解吸(FD)、快原子轟擊(FAB)、電噴霧(ESI)等方法獲得M+H+、M+Na+等準分子離子峰。單糖的鑒定苷鍵全
37、部酸水解后PC,顯色后薄層掃描;苷全甲基化并水解得到甲基化單糖后GC。糖之間的連接位置苷全甲基化甲醇解,13C-NMR苷化位移。糖鏈連接順序緩和酸水解;Smith裂解苷鍵構型酶催化水解法:麥芽糖酶能水解的為-苷鍵,而杏仁苷酶能水解的為-苷鍵。并非所有的-苷鍵都能為杏仁苷酶所水解。分子旋光差法(Klyne法)NMR法1H-NMR:葡萄糖,-苷鍵JH1-H2=68Hz,-苷鍵JH1-H2=34Hz。注意鼠李糖、甘露糖不能用上法鑒別。13C-NMR:1JC1-H1=170Hz(-苷鍵),1JC1-H1=160Hz(-苷鍵)。另外,結構鑒定還可以應用GC(需制備衍生物)、IEC(離子交換色譜法ion
38、exchange chromatography , IEC)、HPLC(折光檢測器)八、苦杏仁苷氰苷,易被酸和酶催化水解。水解所得到的苷元-羥基苯乙腈很不穩定,易分解生成苯甲醛和氫氰酸。 其中苯甲醛具有特殊的香味,通常將此作為鑒別苦杏仁苷的方法。具體操作為:取本品數粒,加水共研,發出苯甲醛的特殊香氣。 苯甲醛可使三硝基苯酚試紙顯磚紅色的反應也可用來鑒定苦杏仁苷的存在。具體操作為:取苦杏仁數粒,搗碎,稱取約0.1g,置試管中,加水數滴使濕潤,試管中懸掛一條三硝基苯酚試紙,用軟木塞塞緊,置溫水浴中,10分鐘后,試紙顯磚紅色。第四章 醌類一、結構與分類苯醌類萘醌類菲醌類對苯醌 鄰苯醌(1,4) (1
39、,2) amphi(2,6) 丹參醌(鄰菲醌)菲醌類蒽醌類單蒽核 丹參新醌(對菲醌)大黃素型R1R2大黃素型(黃色)HCH3大黃酚HCH2OH蘆薈大黃酚OHCH3大黃素OCH3CH3大黃素甲醚HCOOH大黃酸茜草素型R1R2R3茜草素型(橙黃-橙紅)OHHH茜草素OHHOH羥基茜草素OHCOOHOH偽羥基茜草素蒽醌類(單蒽核)氧化蒽酚類蒽醌在堿性溶液中可被鋅粉還原生成氧化蒽酚及其互變異構體蒽二酚,氧化蒽酚及蒽二酚均不穩定蒽酚或蒽酮類蒽醌在酸性溶液中被還原,則生成蒽酚及其互變異構體蒽酮。在新鮮大黃中含有蒽酚類成分,貯存2年以上則檢測不到蒽酚。蒽醌類(雙蒽核)二蒽酮類衍生物:二分子蒽酮脫去一分子氫
40、后相互結合而成的化合物,番瀉苷A、B、C、D。大黃中致瀉的主要成分番瀉苷A,就是因其在腸內轉變為大黃酸蒽酮而發揮作用。二蒽醌類去氫二蒽酮類;日照蒽酮類;中位苯駢二蒽酮類二、理化性質性狀如果無羥基,則無色;隨著助色團酚羥基的引入而表現出一定的顏色。引入的助色團越多,顏色則越深。升華性游離的醌類多具升華性,小分子的苯醌類及萘醌類具有揮發性。溶解性游離醌類多溶于有機溶劑,微溶或不溶于水。而醌類成苷后,極性增大。酸堿性酸性:含COOH含二個以上-OH含一個-OH含二個以上-OH含一個-OH堿梯度萃取法5%碳酸氫鈉溶液5%碳酸鈉溶液1%氫氧化鈉溶液5%氫氧化鈉溶液含COOH或二個-OH含一個-OH含二個
41、或多個-OH含一個-OH顯色反應Feigl反應在堿性條件下加熱與醛類、鄰二硝基苯反應,生成紫色化合物。無色亞甲藍顯色試驗專用于檢識苯醌及萘醌。樣品在白色背景下呈現出藍色斑點Borntragers反應在堿性溶液中,羥基醌類顏色改變并加深,多呈橙、紅、紫紅及藍色。蒽酚、蒽酮、二蒽酮類化合物需氧化形成羥基蒽醌后才能呈色Kesting-Craven反應苯醌及萘醌類的醌環上有未被取代的位置時,在堿性條件下與活性次甲基試劑蒽醌類因不含有未取代的醌環,不發生反應,可用于與苯醌及萘醌類化合物區別。與金屬離子的反應具有-酚羥基或鄰二酚羥基,可與Pb2+、Mg2+等金屬離子形成絡合物。三、提取分離1. 提取:一般
42、選用甲醇、乙醇作為提取溶劑。2. 分離:游離蒽醌衍生物:一般采用溶劑分步結晶法、pH梯度萃取法和色譜法。柱色譜法常用的吸附劑有硅膠、磷酸氫鈣、聚酰胺,一般不用氧化鋁,以免發生不可逆的化學吸附。蒽醌苷類:水溶性較強,需要結合吸附及分配柱色譜分離,常用載體有聚酰胺、硅膠及葡聚糖凝膠。四、結構測定IR蒽醌的羰基頻率未取代蒽醌伸縮頻率為1675cm-1。當蒽醌環上有取代基時:吸電子基團使頻率變高,波數增加,供電子基團使頻率變低,波數減少羥基蒽醌的羰基頻率-OH因與C=O締合,其吸收頻率移至3150cm-1以下;-OH振動頻率較-OH高,在36003150cm-1區間;若只有1個-OH,則大多數在330
43、03390cm-1之間有1個吸收峰;若在36003150cm-1之間有幾個峰,表明蒽醌母核可能有多個-OH。NMR特征是分子離子峰為基峰,游離醌依次脫去兩分子CO,得到M-CO及M-2CO的強峰以及它們的雙電荷峰。五、實例丹參(丹參醌類結構上雖然有菲醌母核,生源上卻屬于二萜類)化學結構脂溶性成分:丹參酮、丹參酮A、丹參酮B、隱丹參酮水溶性成分:丹參素、丹參酸甲、乙、丙鑒定(脂溶性成分)取少量樣品,加濃硫酸2滴,丹參醌顯綠色,隱丹參醌顯棕色,丹參醌I顯藍色。第五章 香豆素和木脂素一、香豆素結構與分類母核簡單香豆素呋喃香豆素苯駢-吡喃酮傘形花內酯補骨脂內酯(6,7-呋喃香豆素、線型)白芷內酯(7,
44、8-呋喃香豆素角型)吡喃香豆素異香豆素其他香豆素類花椒內酯(6,7-吡喃香豆素) 邪蒿內酯(7,8吡喃香豆素)茵陳炔內酯-吡喃酮環上有取代基的香豆素,C-3、C-4上常有苯基、羥基、異戊烯基等取代,這類是指如沙葛內酯、黃檀內酯等。二、香豆素理化性質性狀游離的香豆素多數有較好的結晶,且大多有香味。分子量小的有揮發性,能隨水蒸氣蒸餾,并能升華。香豆素苷多數無香味和揮發性,也不能升華。溶解性游離的香豆素能溶于沸水,難溶于冷水,易溶于甲醇、乙醇、氯仿和乙醚;香豆素苷類能溶于水、甲醇和乙醇,難溶于乙醚等極性小的有機溶劑。熒光性質香豆素母體本身無熒光,而羥基香豆素在紫外光下多顯出藍色熒光,在堿溶液中熒光更
45、為顯著。一般在C-7位引入羥基即有強烈的藍色熒光,加堿后可變為綠色熒光;但在C-8位再引入一羥基,則熒光減至極弱,甚至不顯熒光。呋喃香豆素多顯藍色熒光,熒光性質常用于色譜法檢識香豆素。與堿作用具有內酯環,在熱稀堿溶液中內酯環可以開環生成順鄰羥基桂皮酸鹽,加酸又可重新閉環成為原來的內酯。但長時間在堿中放置或UV光照射,則可轉變為穩定的反鄰羥基桂皮酸鹽,再加酸就不能環合成內酯環。7位甲氧基香豆素較難開環。根據此性質,可利用堿溶酸沉法提取香豆素。顯色反應異羥肟酸鐵反應內酯環在堿性條件下開環,與鹽酸羥胺縮合成異羥肟酸,在酸性條件下與三價鐵離子絡合成鹽而顯紅色。三氯化鐵反應具有酚羥基的香豆素類可與三氯化
46、鐵試劑產生顯色反應,通常為藍綠色。Gibbs反應2,6-二氯(溴)苯醌氯亞胺,在弱堿性條件下可與酚羥基對位的活潑氫縮合成藍色化合物。Emerson反應氨基安替比林和鐵氰化鉀,可與酚羥基對位的活潑氫生成紅色縮合物。區別7,8-呋喃香豆素和6,7-呋喃香豆素。Gibbs反應和Emerson反應都要求必須有游離的酚羥基,且酚羥基的對位要無取代才顯陽性。酚羥基的對位即6-位四、香豆素波譜規律UV300nm處可有最大吸收,峰位置與取代基有關;未取代的香豆素一般有275、284、310三峰。若有羥基取代,尤其是6、7位,則其主要吸收峰紅移,有時幾乎并成一峰。堿液中吸收峰顯著紅移。7-羥基香豆素的max32
47、5nm(4.15),在堿液中即紅移至372nm(4.23)。IR-吡喃酮17451715cm-1處的羰基特征吸收峰;芳環雙鍵的16451625cm-1吸收峰;如果有羥基取代還有36003200cm-1的羥基特征吸收峰。1H-NMRH-3和H-4約在兩組二重峰(J值約為9Hz),化學位移值(H-3:6.16.4,H-4:7.58.3)多數香豆素C-7位有氧取代,苯環上的其余3個芳質子,H-5呈d峰,7.38,J值為9Hz;H-6和H-8在較高場處,6.87,2H,m峰。芳香環上的甲氧基信號一般出現在3.84.0。MS三、香豆素提取與分離水蒸氣蒸餾法小分子的香豆素類因具有揮發性,可采用水蒸氣蒸餾法
48、進行提取。堿溶酸沉法0.5%氫氧化鈉水溶液(或醇溶液)加熱提取,提取液冷卻后再用乙醚除去雜質,然后加酸調節pH至中性,適當濃縮,再酸化。不可長時間加熱,加熱溫度不能過高,堿濃度不宜過大,以免破壞內酯環。系統溶劑法常用石油醚、乙醚、乙酸乙酯、丙酮和甲醇順次萃取。色譜方法吸附劑可用中性和酸性氧化鋁以及硅膠,堿性氧化鋁慎用。常用己烷和乙醚,已烷和乙酸乙酯等混合溶劑洗脫五、木脂素木脂素類多數是游離的,也有少量與糖結合成苷而存在,由于較廣泛地存在于植物的木部和樹脂中,或開始析出時呈樹脂狀,故稱為木脂素。木脂素多數為無色或白色結晶,但新木脂素不易結晶。木脂素多數不揮發,少數如去甲二氫愈創酸能升華,游離木脂
49、素偏親脂性,難溶于水,能溶于苯、氯仿、乙醚、乙醇等。與糖結合成苷者水溶性增大,并易被酶或酸水解。六、藥材實例1. 秦皮:原植物主要有兩種,即木犀科植物大葉白蠟樹及白蠟樹,大葉白蠟樹皮中主要含七葉內酯和七葉苷,而白蠟樹皮中主要含白蠟素和七葉內酯以及白蠟樹苷。 2. 前胡:主要化學成分為多種類型的香豆素及其糖苷、三萜糖苷、甾體糖苷、揮發油等。各種類型的香豆素化合物是前胡的主要代表成分和主要生理活性成分,其中白花前胡以角型二氫吡喃香豆素類為主,紫花前胡以線型二氫呋喃和二氫吡喃香豆素類為主。3.腫節風(草珊瑚):全草含有酚類、鞣質、黃酮苷、香豆素和內酯類化合物。其中香豆素類主要包括異秦皮啶(isofr
50、axidin)、東莨菪內酯(scopoletin)等。4. 補骨脂:有多種香豆素類成分,包括補骨脂內酯(呋喃駢香豆素)、異補骨脂內酯(異呋喃駢香豆素)和補骨脂次素等。5. 五味子:含木脂素較多約為5%,從其果實中分得了一系列聯苯環辛烯型木脂素,主要五味子酯甲、乙、丙、丁和戊。6. 厚樸:厚樸皮中分得了與苯環相連的新木脂素,如厚樸酚以及和厚樸酚。 和厚樸酚 厚樸酚 五味子醇甲 異補骨脂內酯 白花前胡丙素 紫花前胡素C- 白蠟素 R=H 七葉內酯 R=H 白蠟樹苷 R=glc 七葉苷 R=glc第六章 黃酮一、結構與分類基本母核為2-苯基色原酮,基本的碳架為C6-C3-C6。1. 苷元的結構分為:
51、黃酮類、黃酮醇類、二氫黃酮類、異黃酮類、魚藤黃酮類、查爾酮類、二氫黃酮醇類、花色素類、二氫查爾酮類、橙酮類、黃烷-3-醇類、黃烷-3,4-二醇類、雙苯吡酮類、高異黃酮類2-苯基色原酮 C6-C3-C6 黃酮醇 黃酮 二氫黃酮醇類 二氫黃酮醇類 異黃酮類 查爾酮類 花色素 橙酮二、常用性質形態多為結晶性固體,少數為無定形粉末(苷)。旋光性游離苷元中,除二氫黃酮、二氫黃酮醇、黃烷及黃烷醇外,其余均無光學活性。苷類多為左旋、顏色與是否存在交叉共軛體系及助色團(-OH、-OCH3等)的種類、數目、位置(尤其7及4-位顏色加深)有關。黃酮、黃酮醇及其苷類:多顯灰黃黃色。查耳酮:黃橙黃色。二氫黃酮、二氫黃
52、酮醇及黃烷醇:幾乎為無色(交叉共軛體系中斷)。異黃酮:顯微黃色(B環接在3位,缺少完整的交叉共軛體系)。溶解性黃酮、黃酮醇、查耳酮等平面性強的分子,分子間作用力較大,難溶于水;二氫黃酮(醇)、二氫查爾酮類等,系非平面性分子,分子間作用力較小,有利于水分子進入,溶解度稍大。花色苷元(花青素)類雖也為平面性結構,但因以離子形式存在,具有鹽的通性,故親水性較強。黃酮類苷元分子中引入羥基,將增加在水中的溶解度;而羥基經甲基化后,則增加在有機溶劑中的溶解度。酸性酚羥基酸性強弱順序:7,4-二羥基7或4-羥基一般酚羥基5-羥基堿性-吡喃酮環上的醚氧原子,有未共用的電子對,故表現有微弱的堿性,可與強無機酸生成鹽,極不穩定,遇水即可分解。(黃酮類化合物溶于濃硫酸中生成的鹽,常常表現出特殊的顏
溫馨提示
- 1. 本站所有資源如無特殊說明,都需要本地電腦安裝OFFICE2007和PDF閱讀器。圖紙軟件為CAD,CAXA,PROE,UG,SolidWorks等.壓縮文件請下載最新的WinRAR軟件解壓。
- 2. 本站的文檔不包含任何第三方提供的附件圖紙等,如果需要附件,請聯系上傳者。文件的所有權益歸上傳用戶所有。
- 3. 本站RAR壓縮包中若帶圖紙,網頁內容里面會有圖紙預覽,若沒有圖紙預覽就沒有圖紙。
- 4. 未經權益所有人同意不得將文件中的內容挪作商業或盈利用途。
- 5. 人人文庫網僅提供信息存儲空間,僅對用戶上傳內容的表現方式做保護處理,對用戶上傳分享的文檔內容本身不做任何修改或編輯,并不能對任何下載內容負責。
- 6. 下載文件中如有侵權或不適當內容,請與我們聯系,我們立即糾正。
- 7. 本站不保證下載資源的準確性、安全性和完整性, 同時也不承擔用戶因使用這些下載資源對自己和他人造成任何形式的傷害或損失。
最新文檔
- 助理廣告師考試消費市場趨勢分析試題及答案
- 太原社區面試題及答案
- 全科醫學試題及答案詳解
- 地理西亞測試題及答案
- 2024年國際商業設計師考試備考要點試題及答案
- 助理廣告師考試數據分析基礎試題及答案
- c語言測試試題及答案
- 商業設計師考試全新試題及答案揭曉
- 2024年職稱考試紡織品檢驗問答試題及答案
- 破解國際商業美術設計師考試難題試題及答案
- 《電磁感應原理解析》課件
- 成都輸液劑項目可行性研究報告參考范文
- 2025年二級注冊建筑師資格考試《建筑結構、建筑物理與設備》真題卷(附答案)
- 鋰電池基礎知識培訓課件
- 2025-2030城市燃氣產業行業市場現狀供需分析及投資評估規劃分析研究報告
- 緊固件制造企業ESG實踐與創新戰略研究報告
- 2024年陜西高中學業水平合格考試化學試卷真題(含答案詳解)
- 2025年全國國家版圖知識競賽題庫及答案(中小學組)
- 國家義務教育質量監測學生相關因素調查問卷
- smt首件檢驗記錄表
- 青島中瑞泰豐新材料有限公司2萬噸無機環保新材料來料加工項目 環境影響報告書
評論
0/150
提交評論